3.2. Snakemake Workflow to Perform Simulations & Analysis¶
Snakemake was used to develop a simulation and analysis pipeline which automatically simulates the MacroConf dataset. To use this pipeline, we need to specify the compounds, simulation methods, and parameters in a tabular configuration file. Every parameter set is then labelled with a unique hash which is derived from and identifies a specific set of parameters (Fig. 3.3). Based on these parameter sets, requested compounds are parametrised from their SMILES string via the multi-step process described in Fig. 3.2. A cascade of different energy minimization-, equilibration- and simulation-steps are automatically setup, executed, and analysed based on the tabular configuration.
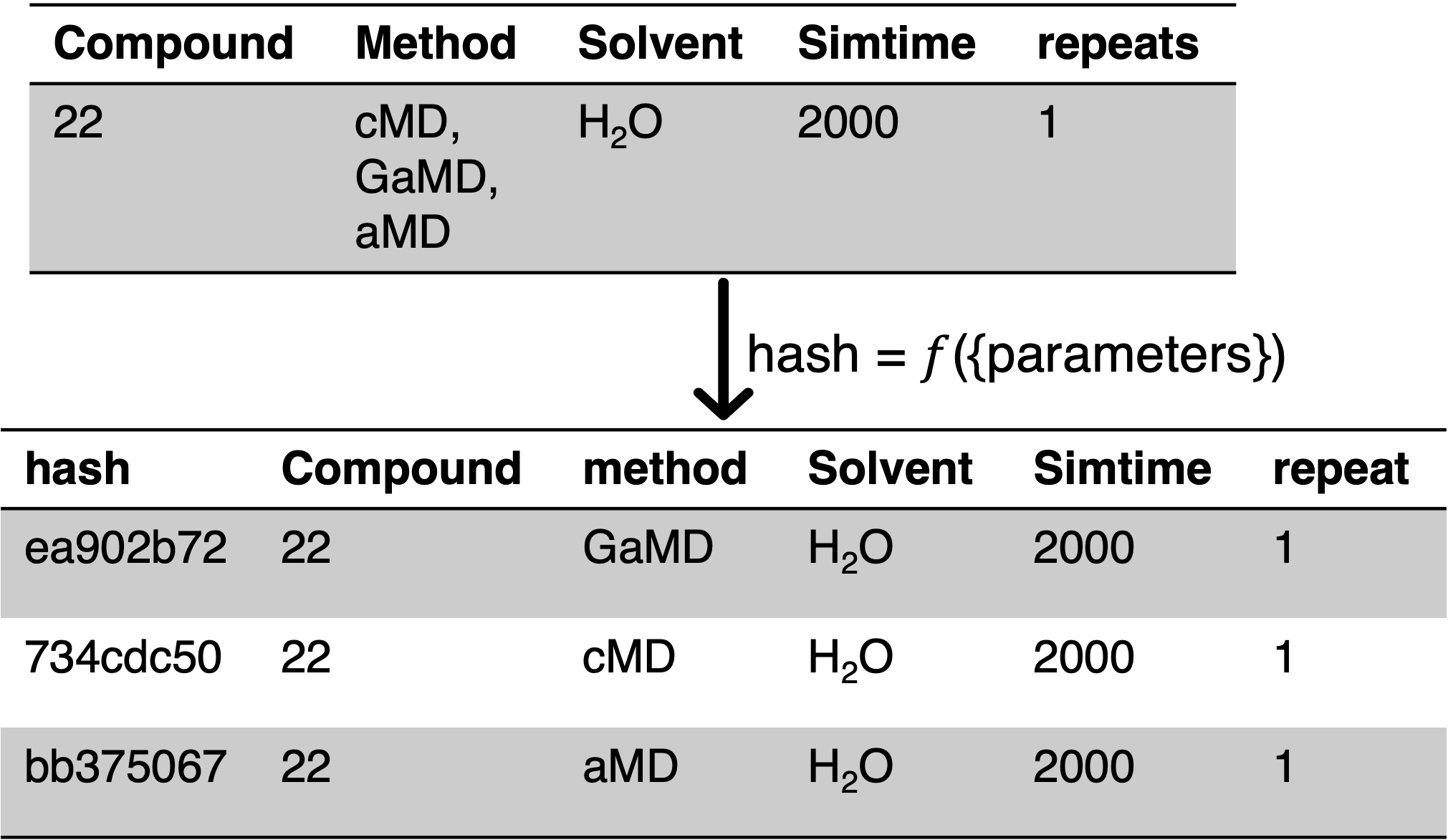
Fig. 3.3 Top: example tabular input for the MacroConf workflow. For convenience, multiple different parameters can be set in the same row. Bottom: as part of the workflow, a hash for every distinct parameter set / simulation run is computed.¶
A detailed description of the entire workflow goes beyond this report, but an example DAG to produce all possible analysis results of the configuration examples shown in Fig. 3.3 are presented in Fig. 3.4. The analysis steps (rules: md_GaMD_anal, md_aMD_anal, md_cMD_anal) will be described in more detail later. Finally, if specified in a separate configuration file, multiple simulations of the same compound with different parameters and simulation methods can be compared with one another (rule: md_comp_analysis). In addition, we can invoke runs of the chemical conformer generators OMEGA or RDKit (with parameters of choice) to compare to the MD simulations.

Fig. 3.4 Directed Acyclic Graph produced via Snakemake, showing how different rules connect to setup, simulate, and analyse MD and conformer generator runs.¶